
BLAST
В биоинформатике BLAST (англ. basic local alignment search tool)[12] — алгоритм и программа для сравнения первичной биологической последовательности, такой как аминокислотные последовательности белков, нуклеотидов ДНК и/или РНК. Поиск с помощью BLAST позволяет исследователю сравнить исследуемую белковую или нуклеотидную последовательность (называемую запросом) с библиотекой или базой данных последовательностей и выявить те, которые схожи с запросом выше определённого порога. Например, после открытия ранее неизвестного гена у мыши, учёный обычно выполняет поиск BLAST по геному человека, чтобы выяснить, есть ли у человека аналогичный ген; BLAST определяет последовательности в геноме человека, схожие с мышиным геном по степени сходства последовательностей.
Инструмент был разработан Стивеном Альтшулем, Уорреном Гишем, Уэббом Миллером, Юджином Майерсом и Дэвидом Липманом в Национальном центре биотехнологической информации (NCBI), который и сегодня продолжает его развитие. Оригинальная научная статья 1990 года, описывающая алгоритм, является одной из самых цитируемых работ в истории науки, насчитывая более 120 000 цитирований[13]. Современная реализация, известная как пакет программ BLAST+, была представлена в 2009 году[14]; по состоянию на июль 2025 года актуальной версией является 2.17.0.
Общие сведения
| BLAST | |||
|---|---|---|---|
| Тип | Инструмент биоинформатики | ||
| Авторы | Стивен Алтшул, Уоррен Гиш, Уэбб Миллер, Юджин Майерс, Дэвид Липман | ||
| Разработчик | NCBI | ||
| Написана на | C и C++[1] | ||
| Интерфейс | Веб-интерфейс, интерфейс командной строки, графический[2] | ||
| Движок | собственный алгоритм[3] | ||
| Операционные системы | UNIX, Linux, Mac, MS-Windows | ||
| Языки интерфейса | Английский[4] | ||
| Первый выпуск | 5 октября 1990[5] | ||
| Аппаратные платформы | x86-64, GPU, FPGA[6] | ||
| Последняя версия | 2.16.0+[7] (25 июня 2024[7]) | ||
| Репозиторий | [8] ftp.ncbi.nlm.nih.gov/bla… | ||
| |||
| |||
| Состояние | активное[11] | ||
| Лицензия | Общественное достояние | ||
| Сайт | blast.ncbi.nlm.nih.gov/Blast.cgi | ||
Предпосылки
BLAST — одна из наиболее широко используемых программ в биоинформатике для поиска по последовательностям[15]. Она решает фундаментальную задачу в биоинформатических исследованиях. Используемый эвристический алгоритм работает быстрее при масштабных поисках по сравнению с методами типа Смита—Ватермана. Такой акцент на скорость критически важен для практического применения алгоритма на огромных геномных базах данных, хотя впоследствии появились ещё более быстрые алгоритмы.
Программа BLAST была разработана в Национальных институтах здравоохранения США (NIH) и основана на стохастическом подходе, разработанном Самуэлем Карлином и Стивеном Алтшулом[16]. Их работа стала «статистической основой BLAST»[17].
- 1990: 5 октября в Journal of Molecular Biology была опубликована основополагающая статья «Basic Local Alignment Search Tool», авторами которой выступили Стивен Алтшул, Уоррен Гиш, Уэбб Миллер, Юджин Майерс и Дэвид Липман. Алгоритм предложил новый эвристический подход, который был значительно быстрее существовавших аналогов и позволял оценивать статистическую значимость найденных совпадений[18]. Эта работа стала одной из самых цитируемых в истории науки, набрав более 120 000 цитирований по данным Google Scholar.
- 1992: Стивен и Джорджия Хеникофф опубликовали новый тип матриц замен аминокислот — BLOSUM (BLOcks SUbstitution Matrix)[19]. Матрица BLOSUM62, представленная в этой работе, впоследствии стала использоваться по умолчанию в большинстве белковых поисков BLAST (например, blastp), значительно повысив их чувствительность[20].
- 1993: Статистическая теория Карлина и Алтшула была расширена для учёта множественных слабых совпадений между последовательностями, что позволило более надёжно идентифицировать отдалённые гомологии[21]. В том же году NCBI представил «Network Entrez» — клиент-серверную версию системы Entrez, сделав её доступной через интернет[22].
- 1994: В журнале Nature Genetics вышла влиятельная статья, в которой рассматривались ключевые проблемы поиска, включая выбор матриц замен и необходимость маскирования участков с низкой сложностью[23].
- 1995: На конференции ISMB-95 была представлена новая версия алгоритма, включавшая два ключевых усовершенствования: Gapped BLAST (поиск с учётом пропусков) и методологию итеративного поиска, которая легла в основу PSI-BLAST[24].
- 1996: Алгоритм Gapped BLAST был подробно описан в публикации Алтшула и Гиша. Новая версия работала примерно в три раза быстрее оригинальной[25].
- 1997: В журнале Nucleic Acids Research вышла статья, официально представившая Gapped BLAST и PSI-BLAST (Position-Specific Iterated BLAST). PSI-BLAST, итеративный метод поиска, значительно повысил чувствительность к слабым, но биологически значимым сходствам[25].
- 1999: Был представлен инструмент «BLAST 2 Sequences» для попарного выравнивания двух последовательностей[26]. Внедрена композиционно-статистическая корректировка, повысившая точность результатов[27].
- 2002: Apple в сотрудничестве с Genentech выпустила Apple/Genentech BLAST — реализацию, оптимизированную для процессоров PowerPC G4 и работавшую до пяти раз быстрее стандартной[28]. В апреле NCBI выпустил BLAST 2.2.3, в котором формат баз данных v4 стал использоваться по умолчанию[27].
- 2003: Издательство O’Reilly опубликовало руководство «BLAST», что свидетельствовало о зрелости и широком распространении инструмента[29]. NCBI усовершенствовал утилиту formatdb[30].
- 2004: Веб-интерфейс BLAST на сайте NCBI был существенно переработан для повышения удобства использования. Появился «BLAST Program Selection Guide» для помощи пользователям в выборе стратегий поиска[31].
- 2006: В веб-интерфейс были добавлены новые функции, такие как подсветка несовпадений и отображение кодирующих регионов (CDS feature)[32].
- 2008: Началась подготовка к выпуску нового поколения программ — BLAST+. Была создана первая версия руководства пользователя[33]. В ноябре NCBI запустил новый веб-сервис Primer-BLAST для разработки специфичных праймеров для ПЦР[34].
- 2009: Состоялся официальный выпуск пакета программ BLAST+, написанного на C++ и заменившего предыдущий «legacy»-набор инструментов (например, blastall).
- 2010: Год стабилизации BLAST+. Вышли релизы 2.2.23 и 2.2.24, направленные на исправление ошибок.
- 2011: Вышел BLAST+ 2.2.26, добавивший поддержку «жёсткого маскирования» (hard-masking) и улучшивший производительность утилиты makeblastdb.
- 2012: Разработан и внедрён новый, более чувствительный метод поиска DELTA-BLAST (Domain Enhanced Lookup Time Accelerated BLAST)[35]. Его поддержка была добавлена в версии BLAST+ 2.2.27.
- 2013: Версия BLAST+ 2.2.28 добавила поддержку форматов вывода JSON и BLAST-XML2.
- 2014: Вышли версии BLAST+ 2.2.29 и 2.2.30 с улучшениями производительности и исправлениями ошибок.
- 2015: Релизы BLAST+ 2.2.31 и 2.3.0 добавили новые опции для PSI-BLAST и бета-версию поддержки формата SAM. Была описана интеграция BLAST+ в биоинформатическую платформу Galaxy[36].
- 2016: Вышел BLAST+ 2.5.0 с улучшениями многопоточности для TBLASTN.
- 2017: Вышли версии 2.6.0 и 2.7.1, которые ускорили поиск по таксономии и улучшили использование многопоточности при нехватке оперативной памяти[37].
- 2019: Версия BLAST+ 2.10.0 улучшила стабильность композиционно-статистической коррекции и значительно (более чем на 20 %) ускорила «быстрые» задачи (например, blastp-fast). NCBI анонсировал переход на новый формат баз данных BLASTDBv5[38].
- 2020: Версия BLAST+ 2.11.0 добавила многопоточность для rpsblast/rpstblastn. В веб-интерфейс были добавлены столбцы с таксономической информацией[39].
- 2021: Версия BLAST+ 2.12.0 представила серьёзную реструктуризацию модуля чтения баз данных, что повысило эффективность многопоточных поисков[40].
- 2022: Вышел BLAST+ 2.13.0, добавивший возможность прямого поиска по базам данных SRA и WGS, а также поддержку архитектуры ARM для Linux[41]. Был запущен облачный инструмент ElasticBLAST и представлена база данных ClusteredNR[42].
- 2023: Версия BLAST+ 2.15.0 упростила таксономический фильтр и ввела автоматический выбор оптимального режима многопоточности, что ускорило поиск по небольшим базам данных в 2-10 раз[43].
- 2024: 25 июня вышла версия BLAST+ 2.16.0, в которой появились опции blastp-fast и blastx-fast для ускоренного поиска. Была представлена новая, более компактная нуклеотидная база данных core_nt, которая в августе стала стандартной для веб-поиска[44].
- 2025: 22 июля вышла версия BLAST+ 2.17.0, добавившая поддержку сжатых файлов FASTA (gzip, bzip2, zstd) в makeblastdb и новый формат вывода CSV с заголовками[45]. В августе база данных ClusteredNR стала стандартной для поиска по белковым последовательностям (Protein BLAST)[42].
Хотя BLAST работает быстрее, чем любая реализация алгоритма Смита—Ватермана в большинстве случаев, он не может «гарантировать оптимальное выравнивание запроса и последовательностей базы данных», как это делает алгоритм Смита—Ватермана. Алгоритм Смита—Ватермана был развитием предыдущего оптимального метода — алгоритм Нидлмана—Вунша, который первым гарантировал нахождение наилучшего возможного выравнивания. Однако требования к времени и памяти у этих оптимальных алгоритмов значительно выше, чем у BLAST.
BLAST эффективнее по времени, чем FASTA, поскольку ищет только наиболее значимые паттерны в последовательностях, при этом обладая сопоставимой чувствительностью. Это становится понятнее при рассмотрении алгоритма BLAST, приведённого ниже.
Для ускорения вычислений BLAST, особенно при работе с большими объёмами данных, используются специализированные аппаратные платформы. Реализации, использующие графические процессоры (GPU), такие как GPU-BLAST, могут значительно повысить скорость поиска за счёт массового параллелизма. Другим подходом является использование программируемых пользователем вентильных матриц (FPGA), на которых создаются специализированные электронные схемы для выполнения наиболее вычислительно затратных этапов алгоритма[46].
Примеры других вопросов, на которые исследователи используют BLAST:
- Какие бактериальные виды имеют белок, родственный по происхождению определённому белку с известной аминокислотной последовательностью
- Какие ещё гены кодируют белки, обладающие структурами или мотивами, подобными только что определённым
BLAST также часто используется как часть других алгоритмов, требующих приближённого сопоставления последовательностей.
BLAST доступен в интернете на сайте NCBI. Существуют различные типы BLAST в зависимости от типа запроса и целевой базы данных. Альтернативные реализации включают:
- AB-BLAST (ранее WU-BLAST) — активно развиваемый коммерческий продукт, являющийся преемником WU-BLAST 2.0[47].
- FSA-BLAST (последнее обновление 12 марта 2013 года)[48].
- ScalaBLAST — высокопроизводительная реализация, последняя значимая версия которой (2.0) вышла в 2013 году, после чего активная разработка не велась[49].
Входные данные для BLAST включают последовательность-запрос, базу данных для поиска и другие опциональные параметры, такие как матрица замен. Последовательность-запрос может быть представлена в нескольких форматах, при этом стандартным и наиболее распространённым является формат FASTA. Программа автоматически определяет формат входных данных. Помимо FASTA, поддерживаются:
- «Голая» последовательность (англ. bare sequence) — последовательность нуклеотидов или аминокислот без строки-описания[50].
- Идентификаторы последовательностей — программа может принимать идентификаторы из баз данных NCBI, такие как номер доступа (англ. accession number) или GI-номер[50].
- Формат GenBank — BLAST может обрабатывать данные, скопированные непосредственно из записей в формате GenBank или GenPept[50].
BLAST не распознаёт в качестве входных данных названия генов, символы или названия белков[50].
Результаты BLAST могут быть представлены в различных форматах, выбор которых зависит от того, предназначен ли результат для чтения человеком или для дальнейшей компьютерной обработки[51]. В командной версии BLAST+ управление форматом вывода осуществляется с помощью параметра -outfmt. Основные форматы включают:
- HTML — формат по умолчанию для веб-версии NCBI. Включает графическое представление, таблицы и детальные выравнивания с гиперссылками[52].
- Текстовый отчёт (англ. Pairwise, `-outfmt 0`) — стандартный, удобочитаемый формат, который подробно отображает выравнивания между запросом и найденными совпадениями[52].
- XML (`-outfmt 5`) — структурированный формат, который легко поддаётся парсингу и удобен для интеграции в автоматизированные конвейеры анализа данных[52]. Версия BLAST+ 2.2.28 (2013 год) добавила поддержку формата BLAST-XML2.
- JSON — формат, поддержка которого была добавлена в версии BLAST+ 2.2.28 (2013 год).
- Табличный формат (англ. Tabular) — один из наиболее часто используемых форматов для последующего анализа. Каждая строка представляет собой одно совпадение (англ. High-scoring Segment Pair, HSP)[53]. Стандартный табличный формат, разделённый знаками табуляции (англ. TSV), задаётся параметром `-outfmt 6`[54]. Существует также вариант с комментариями (`-outfmt 7`), который включает строки с заголовками столбцов[53].
- CSV (англ. Comma-separated values) — представляет ту же табличную информацию, но с запятыми в качестве разделителей (`-outfmt 10`)[53]. Начиная с версии 2.17.0 (2025 год), доступен также формат CSV с заголовками.
- Архивный формат BLAST (англ. ASN.1, `-outfmt 11`) — бинарный формат, который можно впоследствии быстро преобразовать в любой другой с помощью утилиты `blast_formatter`[53].
Процесс
Используя эвристический метод, BLAST находит схожие последовательности, определяя короткие совпадения между двумя последовательностями. Этот процесс называется «seeding». После первого совпадения BLAST начинает строить локальные выравнивания. При поиске сходства последовательностей важны наборы общих букв, называемых словами. Например, если последовательность содержит участок GLKFA, при стандартных условиях размер слова будет 3 буквы. В этом случае слова будут: GLK, LKF и KFA. Эвристика BLAST находит все общие трёхбуквенные слова между интересующей последовательностью и последовательностями из базы данных. Этот результат затем используется для построения выравнивания. После формирования слов для интересующей последовательности формируются и остальные слова. Эти слова должны иметь балл не ниже порога T при сравнении с помощью матрицы оценивания.
Одна из часто используемых матриц для поиска BLAST — BLOSUM62[55], хотя оптимальная матрица зависит от степени сходства последовательностей. После формирования слов и соседних слов они сравниваются с последовательностями в базе данных для поиска совпадений. Пороговый балл T определяет, будет ли конкретное слово включено в выравнивание. После seeding выравнивание длиной всего 3 остатка расширяется в обе стороны алгоритмом BLAST. Каждое расширение увеличивает или уменьшает балл выравнивания. Если балл превышает заранее заданный T, выравнивание включается в результаты BLAST. Если балл ниже T, расширение прекращается, и области с плохим выравниванием не попадают в результаты. Повышение порога T уменьшает область поиска, сокращая число соседних слов, но ускоряя процесс BLAST.
Алгоритм
Для запуска BLAST требуется последовательность-запрос и последовательность для поиска (или база данных последовательностей). BLAST находит подстроки в базе данных, схожие с подстроками запроса. Обычно запрос значительно меньше базы данных, например, запрос может содержать тысячу нуклеотидов, а база данных — несколько миллиардов.
Основная идея BLAST — наличие высоко-скоринговых пар сегментов (HSP) в статистически значимом выравнивании. BLAST ищет высоко-скоринговые выравнивания между запросом и последовательностями базы данных, используя эвристический подход, приближённый к алгоритму Смита—Ватермана. Однако полный перебор по Смиту—Ватерману слишком медленный для поиска по большим геномным базам, таким как GenBank. Поэтому BLAST использует эвристику, менее точную, чем алгоритм Смита—Ватермана, но более чем в 50 раз быстрее[56]. Скорость и относительная точность BLAST — одни из технических инноваций программы. Ключевые этапы алгоритма включают фильтрацию низкосложных регионов, поиск высоко-скоринговых слов и статистическую оценку выравниваний.
Обзор алгоритма BLAST (поиск белок-белок):[56]
- Удаление низкосложных регионов или повторов в последовательности запроса.
- «Низкосложный регион» — участок последовательности, состоящий из небольшого числа различных элементов. Такие регионы могут давать высокие баллы, мешая программе находить действительно значимые последовательности, поэтому их следует фильтровать. Регионы помечаются X (для белков) или N (для нуклеиновых кислот) и игнорируются BLAST. Для фильтрации низкосложных регионов используется программа SEG (для белков) и DUST (для ДНК). Для маскировки тандемных повторов в белках применяется XNU.
- Формирование списка k-буквенных слов из последовательности запроса.
- Например, при k=3 формируются слова длиной 3 из белковой последовательности запроса (k обычно 11 для ДНК), последовательно до конца последовательности. Метод показан на рисунке 1.
 Рис. 1. Метод формирования списка k-буквенных слов запроса[57].
Рис. 1. Метод формирования списка k-буквенных слов запроса[57].
- Например, при k=3 формируются слова длиной 3 из белковой последовательности запроса (k обычно 11 для ДНК), последовательно до конца последовательности. Метод показан на рисунке 1.
- Составление списка возможных совпадающих слов.
- Это одно из главных отличий BLAST от FASTA. FASTA учитывает все общие слова в базе и запросе, а BLAST — только высоко-скоринговые. Баллы рассчитываются сравнением слова из шага 2 со всеми трёхбуквенными словами. Используя матрицу замен (матрица замен), для трёхбуквенного слова возможно 20³ вариантов баллов. Например, сравнение PQG с PEG и PQA даёт баллы 15 и 12 по схеме BLOSUM62. Для ДНК: совпадение +5, несовпадение −4 или +2 и −3. Затем применяется пороговое значение T для сокращения числа возможных совпадающих слов. Слова с баллом выше T остаются, остальные отбрасываются. Например, PEG сохраняется, а PQA — нет при T=13.
- Организация оставшихся высоко-скоринговых слов в эффективное дерево поиска.
- Это позволяет быстро сравнивать высоко-скоринговые слова с последовательностями базы данных.
- Повторение шагов 3-4 для каждого k-буквенного слова запроса.
- Сканирование базы данных на точные совпадения с оставшимися высоко-скоринговыми словами.
- Программа BLAST сканирует базу на наличие оставшихся высоко-скоринговых слов, например PEG, для каждой позиции. При точном совпадении оно используется для инициации возможного выравнивания без разрывов между запросом и базой.
- Расширение точных совпадений до высоко-скоринговых пар сегментов (HSP).
- Оригинальный BLAST расширяет выравнивание влево и вправо от позиции совпадения, пока суммарный балл HSP не начнёт уменьшаться. Упрощённый пример на рисунке 2.
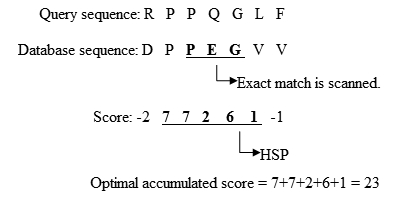 Рис. 2. Процесс расширения точного совпадения. Адаптировано из Biological Sequence Analysis I, Current Topics in Genome Analysis [].
Рис. 2. Процесс расширения точного совпадения. Адаптировано из Biological Sequence Analysis I, Current Topics in Genome Analysis [].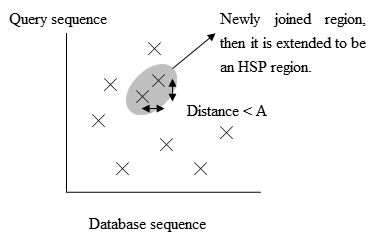 Рис. 3. Позиции точных совпадений.
Рис. 3. Позиции точных совпадений. - Для ускорения разработан BLAST2 (gapped BLAST). BLAST2 использует более низкий порог соседних слов для сохранения чувствительности. Список возможных совпадающих слов становится длиннее. Далее совпадающие регионы, находящиеся на расстоянии A на одной диагонали (рис. 3), объединяются в новый регион. Затем новые регионы расширяются как в оригинальном BLAST, а баллы HSP рассчитываются с помощью матрицы замен.
- Оригинальный BLAST расширяет выравнивание влево и вправо от позиции совпадения, пока суммарный балл HSP не начнёт уменьшаться. Упрощённый пример на рисунке 2.
- Составление списка всех HSP в базе данных с баллом выше порога.
- В список включаются HSP с баллом выше эмпирически определённого порога S. Анализируя распределение баллов выравнивания между случайными последовательностями, выбирается такой порог S, чтобы оставшиеся HSP были статистически значимы.
- Оценка значимости балла HSP.
- BLAST оценивает статистическую значимость каждого балла HSP, используя экстремальное распределение Гумбеля (EVD). (Доказано, что распределение локальных баллов Смита—Ватермана между двумя случайными последовательностями подчиняется EVD Гумбеля. Для локальных выравниваний с разрывами это не доказано.) Вероятность p наблюдать балл S не ниже x вычисляется по формуле
- где
- Статистические параметры и оцениваются по распределению баллов локальных выравниваний без разрывов между запросом и многими перемешанными версиями последовательности базы, подгоняя к EVD Гумбеля. и зависят от матрицы замен, штрафов за разрывы и состава последовательностей (частот букв). и — эффективные длины запроса и базы. Оригинальная длина укорачивается до эффективной для компенсации краевого эффекта. Они вычисляются как
- где — средний ожидаемый балл на пару остатков в выравнивании двух случайных последовательностей. Алтшул и Гиш приводят типичные значения: , , для локального выравнивания без разрывов с BLOSUM62. Использование типовых значений для оценки значимости называется методом lookup table; он неточен. Ожидаемое значение E для совпадения в базе — это число раз, когда несвязанная последовательность базы получит балл S выше x случайно. Ожидание E при поиске по базе из D последовательностей:
- При E можно аппроксимировать распределением Пуассона:
- Это ожидаемое значение или E-value (E-значение) для локального выравнивания без разрывов указывается в результатах BLAST. При объединении HSP (например, при построении выравниваний с разрывами) расчёт модифицируется из-за изменения статистических параметров.
- BLAST оценивает статистическую значимость каждого балла HSP, используя экстремальное распределение Гумбеля (EVD). (Доказано, что распределение локальных баллов Смита—Ватермана между двумя случайными последовательностями подчиняется EVD Гумбеля. Для локальных выравниваний с разрывами это не доказано.) Вероятность p наблюдать балл S не ниже x вычисляется по формуле
- Объединение двух и более HSP в более длинное выравнивание.
- Иногда два и более HSP в одной последовательности базы можно объединить в более длинное выравнивание, что даёт дополнительное подтверждение родства между запросом и базой. Существуют два метода сравнения значимости новых объединённых HSP: метод Пуассона и метод суммы баллов. Например, для пар баллов (65, 40) и (52, 45) метод Пуассона отдаёт предпочтение набору с максимальным меньшим баллом (45>40), а метод суммы баллов — первому набору, так как 65+40=105 > 52+45=97. Оригинальный BLAST использует метод Пуассона; gapped BLAST и WU-BLAST — метод суммы баллов.
- Показ выравниваний с разрывами между запросом и каждой совпавшей последовательностью базы.
- Оригинальный BLAST строит только выравнивания без разрывов, даже если найдено несколько HSP в одной последовательности базы.
- BLAST2 строит единое выравнивание с разрывами, включающее все найденные HSP. При этом для расчёта балла и E-value используются соответствующие штрафы за разрывы.
- Вывод всех совпадений с ожидаемым значением ниже порогового параметра E.


















Существует несколько разновидностей программы BLAST, каждая из которых предназначена для определённого типа задач. Основные программы включают:
- BLASTn (Nucleotide BLAST)
- Сравнивает нуклеотидную последовательность-запрос с нуклеотидной базой данных.
- BLASTp (Protein BLAST)
- Сравнивает аминокислотную последовательность-запрос с белковой базой данных.
- BLASTx
- Сравнивает нуклеотидную последовательность-запрос, транслированную во всех шести возможных рамках считывания, с белковой базой данных. Этот метод полезен для поиска потенциальных белок-кодирующих генов в новой последовательности ДНК.
- tBLASTn
- Сравнивает белковую последовательность-запрос с нуклеотидной базой данных, динамически транслируя последовательности из базы данных во всех шести рамках считывания. Используется для поиска гомологов белка в неаннотированных геномных или транскриптомных данных.
- tBLASTx
- Сравнивает нуклеотидную последовательность-запрос, транслированную в шести рамках, с нуклеотидной базой данных, также транслируемой в шести рамках. Это самый вычислительно затратный метод, который полезен для поиска отдалённых гомологов между нуклеотидными последовательностями.
Помимо основных программ, существует ряд специализированных версий и инструментов:
- PSI-BLAST (Position-Specific Iterated BLAST)
- Итеративный метод для поиска отдалённых гомологов белка. После первого поиска программа создаёт позиционно-специфическую матрицу весов (PSSM) из найденных значимых выравниваний и использует её для следующего, более чувствительного раунда поиска.
- DELTA-BLAST (Domain Enhanced Lookup Time Accelerated BLAST)
- Представленный в 2012 году, этот метод повышает чувствительность поиска белков за счёт предварительного поиска по базе данных консервативных доменов для построения более точной PSSM перед основным поиском.
- BLAST 2 Sequences (bl2seq)
- Инструмент для попарного выравнивания двух последовательностей (белковых или нуклеотидных) с использованием алгоритма BLAST, что позволяет избежать поиска по всей базе данных.
- Primer-BLAST
- Инструмент для дизайна и проверки специфичности праймеров для ПЦР. Он интегрирует программу Primer3 с поиском BLAST для проверки того, что праймеры будут амплифицировать только целевую последовательность.
- IgBLAST
- Специализированный инструмент для анализа последовательностей иммуноглобулинов (антител) и Т-клеточных рецепторов (TCR). Он помогает идентифицировать V(D)J-гены и анализировать соматические гипермутации.
- Magic-BLAST
- Инструмент, оптимизированный для картирования данных секвенирования нового поколения (NGS), таких как прочтения РНК-секвенирования, на референсный геном или транскриптом. Он способен обрабатывать большое количество коротких последовательностей и генерировать вывод в формате SAM[58].
- Поиск по архивам SRA/WGS
- Специализированные версии, такие как blastn_vdb и tblastn_vdb, позволяют выполнять поиск напрямую по базам данных необработанных данных, таким как SRA и Whole Genome Shotgun (WGS), без предварительного форматирования.
Современный пакет программ BLAST+ изначально поддерживает многопоточность, позволяя ускорять поиск на многоядерных процессорах с помощью параметра -num_threads[59]. Однако для обработки сверхбольших объёмов данных и достижения максимальной производительности в высокопроизводительных вычислительных средах (HPC) используются более сложные подходы к параллелизации.
Исторически одним из самых популярных подходов к параллелизации BLAST является использование технологии MPI для распределения задач между узлами кластера. Основные стратегии включают разделение базы данных на фрагменты (сегментацию) или распределение множества запросов между вычислительными узлами. После завершения параллельных вычислений результаты объединяются в единый отчёт[60].
К наиболее известным реализациям относятся mpiBLAST, ScalaBLAST и DCBLAST. mpiBLAST — одна из ранних и наиболее влиятельных параллельных версий, основанная на сегментации базы данных. Проект активно развивался в 2000-х годах: версия 1.3.0 вышла в конце 2004 года и представила механизм «конвейеризации базы данных»[61], а версия 1.4.0 (2006 год) исправила проблемы с производительностью[62]. Последней значимой версией стала 1.6.0, после чего активная разработка прекратилась примерно в 2010—2013 годах[63]. К 2014 году mpiBLAST был основан на устаревшем ядре «legacy» BLAST, в то время как появились новые инструменты, использующие современный BLAST+[64]. Разработка ScalaBLAST также не ведётся; его последняя версия (2.0) вышла в 2013 году. DCBLAST — более современная программная оболочка (wrapper) для NCBI BLAST+, последние изменения в коде которой датируются 2020 годом[65].
Для ускорения наиболее вычислительно затратных этапов алгоритма BLAST используются специализированные аппаратные платформы.
- Графические процессоры (GPU): Благодаря своей архитектуре, ориентированной на массовый параллелизм, GPU эффективно ускоряют поиск. Реализации, такие как GPU-BLAST, используют технологию CUDA для переноса вычислений на графический процессор и могут достигать многократного ускорения по сравнению с многопоточными реализациями на CPU.
- Программируемые пользователем вентильные матрицы (FPGA): Этот подход предполагает создание специализированных электронных схем, «заточённых» под конкретные шаги алгоритма, такие как поиск совпадений (hit finding) или расширение выравниваний (extension)[66]. Реализации на FPGA также демонстрируют значительный прирост производительности по сравнению с программными версиями, работающими на универсальных процессорах.
Программа BLAST предоставляет несколько типов пользовательских интерфейсов, рассчитанных на разные категории пользователей и различные задачи.
- Веб-интерфейс
- Самым популярным способом использования BLAST является веб-интерфейс, предоставляемый NCBI. Этот интерфейс позволяет пользователям вставлять или загружать последовательности, выбирать базы данных для поиска и настраивать параметры через интуитивно понятные веб-формы[67]. Результаты представляются в наглядном графическом и текстовом виде[67], что делает этот вариант идеальным для исследователей, которым необходимо выполнить единичные или нечастые поиски.
- Интерфейс командной строки (CLI)
- Для более продвинутых и массовых задач NCBI распространяет пакет программ BLAST+, который используется через интерфейс командной строки. Этот текстовый интерфейс предоставляет пользователям максимальную гибкость, позволяя проводить массовые поиски, использовать собственные базы данных, а также интегрировать BLAST в сложные вычислительные конвейеры и скрипты[68]. CLI является основным способом работы с BLAST на высокопроизводительных вычислительных кластерах.
- Локальные программы с графическим интерфейсом (GUI)
- Для пользователей, которым необходимо выполнять поиск на локальном компьютере (например, с частными базами данных), но которые не обладают навыками работы в командной строке, существуют сторонние программы с графическим интерфейсом[69]. Эти приложения, такие как BlastGUI и BlastUI, предоставляют оболочку для управления локальными версиями BLAST+, позволяя создавать собственные базы данных, запускать поиск и визуализировать результаты в удобном виде, не требуя знания команд[69].
Пакет программ BLAST+ написан преимущественно на языках программирования C++ и C[70]. Ядро программы представляет собой собственный эвристический алгоритм, который в документации для разработчиков именуется «ядром BLAST» (англ. BLAST engine)[71].
Исходный код находится в общественном достоянии и официально распространяется NCBI через свой FTP-сервер. Самая последняя версия пакета всегда доступна для загрузки в каталоге с псевдонимом «LATEST». Хотя NCBI имеет официальный аккаунт на GitHub, он используется преимущественно для документации (например, репозиторий ncbi/blast_plus_docs), а не для хранения основного исходного кода[72].
Пользовательский интерфейс программы BLAST, включая веб-интерфейс на сайте NCBI и пакет программ для командной строки, доступен только на английском языке. Вся официальная документация, руководства и обучающие материалы также предоставляются на английском языке.
Альтернативы BLAST
Предшественник BLAST — FASTA, также используется для поиска сходства белковых и ДНК-последовательностей. FASTA предоставляет аналогичный набор программ для сравнения белков с белковыми и ДНК-базами, ДНК с ДНК и белковыми базами, а также дополнительные инструменты для работы с короткими пептидами и ДНК. Кроме того, пакет FASTA включает SSEARCH — векторизованную реализацию строгого алгоритма Смита—Ватермана. FASTA медленнее BLAST, но поддерживает больше матриц оценивания, что облегчает настройку поиска под конкретную эволюционную дистанцию.
Инструмент, ранее известный как WU-BLAST (Washington University BLAST), продолжает развитие под названием AB-BLAST (Advanced BioComputing BLAST). Он является коммерческим продуктом и позиционируется как прямой преемник WU-BLAST 2.0[73]. По заявлениям разработчиков, AB-BLAST до двух раз быстрее и более чувствителен, чем NCBI BLAST[73], сохраняя при этом обратную совместимость с параметрами командной строки своего предшественника. Продукт активно поддерживается (версия 3.0 вышла в декабре 2018 года, а обновления выпускались как минимум до марта 2020 года), но для его использования требуется лицензия (существует бесплатная персональная некоммерческая лицензия)[74][75].
Крайне быстрый, но менее чувствительный аналог BLAST — BLAT (Blast Like Alignment Tool). В отличие от линейного поиска BLAST, BLAT использует k-мерное индексирование базы, что позволяет быстрее находить seed-совпадения. На сегодняшний день BLAT считается «унаследованным инструментом», который не находится в активной разработке; последняя известная версия (v.35) была выпущена в 2012 году[76][77]. Ещё одна похожая программа — PatternHunter.
FSA-BLAST — ещё одна альтернативная реализация, которая, по заявлениям разработчиков, работала значительно быстрее оригинального NCBI-BLAST. Однако проект не находится в активной разработке, последнее обновление датируется 12 марта 2013 года[48].
Развитие технологий секвенирования в конце 2000-х сделало поиск очень схожих нуклеотидных совпадений актуальной задачей. Новые программы для выравнивания используют BWT-индексирование базы (обычно генома). Входные последовательности можно быстро отображать, а результат обычно выдаётся в формате BAM. Примеры: BWA, SOAP, Bowtie.
Для идентификации белков, поиска известных доменов (например, из Pfam) путём сопоставления с скрытыми марковскими моделями популярен пакет HMMER.
Альтернатива BLAST для сравнения двух банков последовательностей — PLAST. PLAST — высокопроизводительный инструмент для поиска сходства между банками последовательностей, основанный на алгоритмах PLAST[78] и ORIS[79]. Разработка инструмента неактивна; последняя известная версия (2.3.2) была выпущена в 2017 году[80].
В метагеномике, где требуется сравнивать миллиарды коротких ДНК-ридов с десятками миллионов белковых референсов, активно развивается инструмент DIAMOND[81]. Он работает до 20 000 раз быстрее BLASTX при сохранении высокой чувствительности. Проект активно поддерживается, регулярно выходят новые версии с улучшениями производительности и новыми функциями[82].
Открытое ПО MMseqs2 — ещё одна активно развиваемая альтернатива BLAST/PSI-BLAST, превосходящая существующие инструменты по всему диапазону компромисса скорость/чувствительность. Проект регулярно обновляется; последняя версия (Release 18) вышла в июле 2024 года и включала улучшения поддержки GPU[83][84].
Оптические вычисления рассматриваются как перспективная альтернатива электронным реализациям. Пример — OptCAM, который быстрее BLAST[85].
Визуализация вывода BLAST
Для интерпретации результатов BLAST существует различное ПО. По способу установки, функциям анализа и технологиям доступны следующие инструменты:[86]
- Сервис NCBI BLAST
- Общие интерпретаторы вывода BLAST, с графическим интерфейсом: JAMBLAST, Blast Viewer, BLASTGrabber
- Интегрированные среды BLAST: PLAN, BlastStation-Free, SequenceServer
- Парсеры вывода BLAST: MuSeqBox, Zerg, BioParser, BLAST-Explorer
- Специализированные инструменты: MEGAN, BLAST2GENE, BOV, Circoletto
Одним из популярных инструментов для визуализации и интерактивной работы с результатами является SequenceServer. Он представляет собой веб-интерфейс для запуска локальных поисков BLAST. Версия 2.0, ставшая стабильной в январе 2022 года, представила новую архитектуру и три новые визуализации, включая гистограмму распределения длин совпадений[87][88]. Версия 2.1 (август 2023 года) добавила возможность облачного сохранения результатов по секретной ссылке[88], а крупное обновление 3.0 вышло 14 февраля 2024 года[89]. Актуальной версией по состоянию на июнь 2024 года является 3.1, которая включает интеграцию с BLAST+ 2.15, ускоренный запуск и улучшенную обработку ошибок[88].
Примеры визуализации результатов BLAST показаны на рисунках 4 и 5.


Применение BLAST
BLAST может использоваться для различных целей: идентификация видов, поиск доменов, построение филогении, картирование ДНК и сравнение.
- Идентификация видов
- С помощью BLAST можно корректно определить вид или найти гомологичные виды. Это полезно, например, при работе с ДНК неизвестного вида.
- Поиск доменов
- При работе с белковой последовательностью её можно ввести в BLAST для поиска известных доменов.
- Построение филогении
- На основе результатов BLAST можно построить филогенетическое дерево на веб-странице BLAST. Однако такие деревья менее надёжны, чем построенные специализированными филогенетическими методами, и подходят только для предварительного анализа.
- Картирование ДНК
- При работе с известным видом и поиске гена в неизвестной позиции BLAST позволяет сравнить хромосомное положение интересующей последовательности с релевантными последовательностями в базе. Для этого у NCBI есть инструмент Magic-BLAST[90].
- Сравнение
- При работе с генами BLAST позволяет находить общие гены у двух родственных видов и использовать аннотации одного организма для другого.
- Классификация таксономии
- BLAST может использовать генетические последовательности для сравнения нескольких таксонов с известными таксономическими данными, что позволяет получить картину эволюционных связей между видами (рис. 6). Это полезно для поиска сиротских генов, поскольку если ген обнаруживается вне родственной линии, он не считается сиротским.
- Хотя этот метод полезен, для поиска гомологов более точны парные и множественные выравнивания.

Примечания
Ссылки
- blast.ncbi.nlm.nih.gov/Blast.cgi — официальный сайт BLAST
- BLAST+ executables — бесплатные исходные коды
| В библиографических каталогах |
|---|
