



Реакция Альдера
Реакция Альдера (Alder), также известная как еновая реакция, представляет собой процесс присоединения енофила к алкену через стадию переноса протона из аллильного положения. Химизм реакции можно описать следующим образом: енофил взаимодействует с алкеном, что приводит к образованию нового продукта через промежуточные стадии, включающие перенос протона.[1]
История открытия
Реакция Альдера (еновая реакция) была впервые описана Куртом Альдером (Kurt Alder) и его коллегами Ф. Пашером (F. Pascher) и А. Шмитцем (A. Schmitz) в их основополагающей работе 1943 года (опубликованной в Berichte der Deutschen Chemischen Gesellschaft).[2]
Предпосылки к открытию
К моменту открытия еновой реакции Альдер уже был известен как один из создателей реакции Дильса-Альдера (1928), которая легла в основу многих последующих исследований в области циклоприсоединения. Однако еновая реакция представляла собой принципиально иной механизм, включающий перенос протона и электрофильное присоединение, что расширило понимание реакционной способности алкенов. В 1981 году Вольфганг Оппольцер (Wolfgang Oppolzer) опубликовал обзор (Pure and Applied Chemistry), в котором систематизировал современные на тот момент представления о еновой реакции. Он подчеркнул её важность в синтезе сложных карбоциклических структур и показал, как реакция может протекать в стереоселективном варианте при использовании катализаторов.
Механизм реакции
Механизм еновой реакции[3] включает несколько ключевых стадий:
- Активация енофила, который выступает в роли электрофильного агента.
- Перенос протона из аллильного положения алкена, что приводит к образованию промежуточного карбокатиона.
- Атака енофила на карбокатион с последующим образованием нового связного продукта.

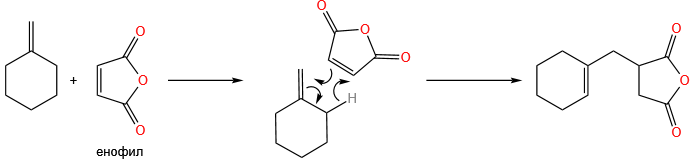{kind=link}
Алкены в этой реакции представляют собой молекулы с π-связями, которые содержат по меньшей мере один атом водорода в аллильном, пропаргическом или α-положении. Возможные компоненты белков включают олефиновые, ацетиленовые, алленовые, ароматические, циклопропильные связи. Фенол может выступать в качестве енового компонента, например, в реакции с дигидропираном, но для этого требуются высокие температуры (150—170 °C).; тем не менее, конденсированные енолы и циклоалкены подвергаются еновым реакциям при гораздо более низких температурах. Аналогично, реакции Альдера с енолами или енолятами классифицируются как реакции енольной конденсации.
Енофилы, как правило, являются электрофильными олефинами. Также участвует ряд молекул с π-связями и электроноакцепторными заместителями. В дополнение к множественным углерод-углеродным связям (олефины, ацетилены), енофилы могут содержать множественные углерод-гетеро-связи (C=O в случае карбонил-еновых реакций, C=N, C=S, C≡P), гетеро-гетеро-кратные связи (N=N, O=If, N=O, S=O), кумуленовые системы (N=S=O, N=S=N, N=Se=N, C=O, C=C=S, SO2) и заряженные π-системы (C=+, C=S+, C≡O+, C≡N+).
Как и в случае любого циклоприсоединения, успех реакции в значительной степени определяется пространственной доступностью водорода при альфа углероде. Как правило метильные (CH₃-) и метиленовые (CH₂-) атомы водорода отрываются гораздо легче, чем метиновые (CH-). Порядок реакционной способности можно представить следующим образом: первичный (1°) > вторичный (2°) > третичный (3°). Например, в молекуле изопрена (CH₂=C(CH₃)-CH=CH₂) енофил предпочтёт забрать водород от метильной группы CH₃- или от метиленовой группы CH₂-, а не тот, что рядом с двойной связью (метиновый). Этот порядок не всегда зависит от того, насколько устойчивым будет конечный продукт. Это кинетическое предпочтение, то есть реакция просто быстрее идёт по таким атомам.
В термических реакциях наблюдается строгая последовательность: первичные атомы водорода более реакционноспособны, чем вторичные, которые, в свою очередь, активнее третичных. Однако при использовании кислот Льюиса в качестве катализатора это правило может нарушаться, и относительная лёгкость отрыва водорода начинает определяться конкретной парой енофил-катализатор.
Ключ к предсказанию основного продукта реакции — региоизомера — лежит в анализе стабильности переходного состояния. На ранней, асимметричной стадии формирования сигма-связи происходит гетеролитическое разделение зарядов: на атоме углерода ена, отдающем водород, развивается частичный положительный заряд (+δ), а на енофиле, принимающем водород, — частичный отрицательный заряд (-δ). Преимущественным путём реакции становится тот, в котором эти возникающие частичные заряды стабилизированы наилучшим образом. Стабилизация положительного заряда на ене происходит благодаря электронодонорным заместителям, таким как алкильные группы, в то время как отрицательный заряд на енофиле стабилизируется его собственной электронодефицитной природой, позволяющей делокализовать заряд. Таким образом, реакция следует по пути наименьшего электронного напряжения, что и обуславливает её региоселективность.[4]
Примеры применения
Еновая реакция нашла широкое применение в органическом синтезе, некоторые примеры:
- Синтез сложных природных соединений в работе M.Stratakis и M.Orfanopoulos (Tetrahedron, 2000) еновая реакция использовалась для функционализации природных соединений, таких как терпены и стероиды. Реакция позволяла селективно вводить новые функциональные группы в сложные молекулы, сохраняя их каркас. Авторы показали, что еновая реакция может протекать в мягких условиях, что важно для работы с термолабильными соединениями.[5]
- Каталитические асимметрические реакции. В обзоре Mikami и Nakai (Catalytic Asymmetric Synthesis, 2000) обсуждалось применение еновой реакции в стереоконтролируемом синтезе. Разработаны хиральные катализаторы (например, на основе комплексов палладия и родия), позволяющие получать оптически активные продукты с высоким энантиоизбытком (до 99 % ee). Реакция использовалась для синтеза биологически активных молекул, таких как простагландины и аминокислотные производные. Были предложены модели асимметрической индукции, объясняющие высокую стереоселективность процесса.[6]
- Разработку новых методов функционализации алкенов В исследовании Lei, He и Zhang (JACS, 2002) еновая реакция была адаптирована для радикальной функционализации алкенов. Использование фотохимической активации или переходных металлов (например, меди) для генерации радикальных интермедиатов. (Lei, He, Zhang, 2002). Реакция применялась для создания C-C и C-гетероатомных связей (например, в синтезе β-функционализированных карбонильных соединений). Метод показал высокую совместимость с различными функциональными группами, что сделало его полезным для медленно- и позднестадийной модификации сложных молекул.[7]